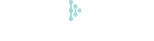


Related products
相关产品

发表文章:
TRACERx analysis identifies a role for FAT1 in regulating chromosomal instability and whole-genome doubling via Hippo signalling
发表期刊:
Nature Cell Biology
原文链接:
https://doi.org/10.1038/s41556-024-01558-w
使用仪器:
NL5+ 深层高速线重扫共聚焦显微镜
一、研究背景
染色体不稳定性(chromosomal instability, CIN)是实体肿瘤,尤其是非小细胞肺癌(non-small cell lung cancer, NSCLC)中普遍存在的现象。CIN 会加剧肿瘤内异质性,促进肿瘤进化,并导致患者预后不良和治疗耐药。全基因组加倍(whole-genome doubling, WGD)是 CIN 的一种极端表现形式,它不仅能够缓冲 CIN 带来的有害效应,还会进一步推动基因组层面的演化。尽管 CIN 和 WGD 在癌症进展中具有重要作用,但 NSCLC 中驱动这两者的具体遗传事件尚不明确。本文作者利用TRACERx 队列的多区域外显子测序数据,系统筛选与 WGD 和 CIN 相关的驱动基因,并重点揭示了 FAT1 基因在其中扮演的关键角色。

图1 来自 TRACERx 100 队列的基因组加倍相关驱动因子的 DDR 和 CIN 功能丧失筛选。说明:a,候选基因选择流程图;b,DDR 和 CIN 筛选设计示意图;c,维恩图显示六个同时参与 DDR 和 CIN 的驱动基因;d,通过 DR-GFP 同源重组报告实验验证六个候选基因;e,通过 Diva U2OS-AsiSI 位点定向切除实验验证六个候选基因;f,箱线图量化 A549 细胞中六个候选基因敲降后 RAD51 焦点形成;g,TRACERx 421 队列中六个候选基因的驱动突变分布和突变时间。FAT1 以红色突出显示。
二、解决的核心问题
本研究旨在回答三个核心问题:
第一,哪些肿瘤驱动基因突变与 NSCLC 中的 WGD 和 CIN 密切相关?
第二,FAT1 基因突变如何影响 DNA 损伤修复和染色体稳定性?
第三,FAT1 是否通过 Hippo 信号通路分别调控 CIN 和 WGD,并进而影响肿瘤进化和治疗耐药?
三、研究方法
研究团队首先分析了 TRACERx 421 名 NSCLC 患者的肿瘤样本,识别出与 WGD 共发生的 37 个肿瘤抑制基因。随后,他们在四种肺癌细胞系中开展了基于 RNA 干扰的高内涵筛选,结合 DNA 双链断裂诱导剂、复制压力诱导剂以及人工染色体(HAC)丢失报告系统,鉴定出六个同时影响 DNA 损伤修复和染色体稳定性的驱动基因,其中 FAT1 因高突变频率和临床相关性被优先研究。在机制层面,研究者采用了 DR-GFP 同源重组报告实验、DIvA 位点特异性 DNA 末端切除和染色体易位检测、免疫荧光分析修复蛋白焦点形成、DNA 纤维实验检测复制叉倒塌、活细胞显微成像追踪有丝分裂错误和胞质分裂失败,以及 TEAD 转录活性报告系统解析 Hippo 通路输出。此外,还利用 CRISPR/Cas9 构建了 FAT1 敲除细胞系,并在奥希替尼耐药模型中评估了 FAT1 缺失对肿瘤进化的影响。

图 2 TRACERx 100 队列中基因组加倍相关驱动因子的 DDR 和 CIN 功能丧失筛选总结。说明:a,在 4 种 LUAD 细胞系中对 37 个 TRACERx 驱动因子进行DDR筛选的无监督聚类结果;b,HAC 实验用于评估基因敲降和轻度复制压力对 CIN 的影响。
四、研究结果
#01 FAT1 突变在肺鳞癌中被正选择且多发生于 WGD 之前
研究发现,FAT1 失活突变在肺鳞癌(LUSC)中更为常见,且 dN/dS 比值显著大于 1,表明存在正选择。进一步的时间分析显示,FAT1 突变多发生在 WGD 事件之前。在 4q35.2 位点,FAT1 拷贝数缺失、启动子高甲基化和表达下调在 WGD 阳性肿瘤中显著富集,提示 FAT1 失活是肿瘤早期的重要事件。

图 3 FAT1 功能丧失在 TRACERx 421 队列中富集,并导致有丝分裂错误率升高和 WGD。说明:a,dN/dS 比值分析示意图及结果,显示 FAT1 截短突变在LUSC肿瘤进化早期被选择;b,仅具有克隆性 WGD 的 TRACERx LUAD 和 LUSC 肿瘤的 GISTIC 分析,显示 FAT1 基因组位点(4q35.2)缺失在克隆性 WGD 肿瘤中被正选择;c,MSAI 分析显示 4 号染色体上 FAT1 基因所在区域在 LUSC 中频繁丢失;d,上图显示 4q35.2 区域基因,下图显示 gnomAD 中针对这些基因缺失的选择压力, FAT1 具有最低的 LOF 耐受性。
#02 FAT1 缺失导致同源重组修复缺陷
在功能上,FAT1 敲低或敲除并不影响 DNA 损伤早期的 ATM、γH2A.X 和 53BP1 焦点形成,但显著抑制了 CtIP、BRCA1 和 RAD51 的焦点形成,表明同源重组(HR)修复的启动和过程受到损害。HR 报告实验和单链 DNA 末端切除实验进一步证实了 HR 效率的下降。有趣的是,FAT1 的 C 端胞内段(HA-FAT1CD)能够部分挽救 RAD51 焦点形成,且该片段具有核定位特征,提示 FAT1 可能通过核内功能参与 HR 修复。

图 4 FAT1 缺失削弱同源重组修复。说明:a,箱线图显示 FAT1 siRNA 敲降对 A549 细胞中早期 DNA 损伤信号及 53BP1 结合的影响;b,FAT1 功能结构域示意图;c,在 FAT1 CRISPR 敲除 A549 细胞中过表达 HA-FAT1CD 可部分恢复 RAD51 焦点形成;d-f,TCGA LUAD 数据中 FAT1 拷贝数改变与 HRD 相关基因组疤痕(LST、TAI、LOH)的相关性,以及 TRACERx LUAD 中 FAT1 驱动突变评分;g,EJ5-GFP 远端连接报告实验显示 FAT1 敲降不影响远端连接;h,EJ2-GFP 替代性末端连接报告实验显示 FAT1 敲降显著降低替代性末端连接效率。
#03 FAT1 缺失同时引发结构性和数值性 CIN
FAT1 缺失导致复制叉塌陷率显著增加,微核、染色质桥、滞后染色体、放射状染色体和染色单体间隙等结构异常明显增多。同时,通过染色体计数和 ImageStream 分析,FAT1 敲低细胞出现明显的染色体数目偏离,即数值性 CIN。在 PC9 及其 WGD 六倍体克隆中,FAT1 缺失进一步加剧了有丝分裂错误和复制压力。

图 5 FAT1 缺失增加复制压力和微核。说明:a,FAT1 敲除加剧 A549 细胞中复制叉停滞;b,TCGA LUAD 中 FAT1 拷贝数缺失与加权基因组不稳定性指数显著相关;c,Genomics England LUAD 和 LUSC 队列中 FAT1 野生型与突变型肿瘤的 indel 数量比较;d,FAT1 siRNA 敲降诱导 cyclin A 阴性 U2OS 细胞中 53BP1 小体形成;e,f,FAT1 敲降诱导 U2OS 细胞微核形成,包括着丝粒性和无着丝粒微核;g,h,A549 和 T2P 细胞中 FAT1 缺失在复制压力下增加微核率。
#04 FAT1 缺失通过胞质分裂失败驱动 WGD
通过活细胞显微成像,研究者发现 FAT1 缺失并不会增加有丝分裂失败或内复制,而是显著提高了胞质分裂失败(cytokinesis failure)的频率,从而直接导致 WGD。EdU 掺入和 MCM7 加载实验也证实,FAT1 敲除细胞中出现更多大于 6N DNA 含量的复制群体。

图 6 FAT1 缺失导致有丝分裂错误率升高并引起 WGD。说明:a,PC9 细胞中 FAT1 敲除后 EdU 掺入检测 WGD 的代表性点图;b,Western blot 验证 FAT1 敲除及 EdU 掺入定量,显示 FAT1 敲除显著增加 WGD 群体;c,活细胞成像显示 FAT1 敲降不通过有丝分裂旁路促进 WGD;d,FAT1 敲降显著增加胞质分裂失败率;e,FAT1 敲降增加正常有丝分裂后子细胞核形变异常率。
#05 FAT1 通过 Hippo–YAP1 轴调控 WGD,但 HR 缺陷不依赖 YAP1
在信号通路层面,FAT1 缺失导致 YAP1 核定位增加和 TEAD 转录活性上升,但并不影响 MEK-ERK 或 Yes 激酶。使用 TEAD 报告系统发现,FAT1 的跨膜区及 MIB2 相互作用域对于抑制 YAP1 活性是必需的。值得注意的是,尽管 FAT1/YAP1 共敲除可以完全逆转胞质分裂失败和 WGD,却无法恢复 HR 缺陷和结构 CIN。相反,组成性活化的 YAP1 突变体(YAP1ssA)足以诱导 WGD。这些结果表明,FAT1 通过两条独立的下游通路发挥作用:YAP1 依赖的通路导致 WGD 和数值性 CIN,而 YAP1 非依赖的通路导致HR缺陷和结构性 CIN。

图 7 FAT1 和 YAP1 共敲除逆转胞质分裂失败但不逆转 HR 缺陷。说明:a,DR-GFP 实验显示 FAT1/YAP1 共敲除不恢复 HR 效率;b,ssDNA 切除实验显示共敲除不恢复末端切除;c,RAD51 焦点形成显示共敲除不恢复 HR;d,e,有丝分裂错误率量化及代表图显示共敲除不拯救染色体桥和 FANCD2 标记的染色质桥;f,活细胞成像显示共敲除完全拯救胞质分裂失败和 WGD(左),部分拯救核形变(右);g,YAP1SSA 过表达在 TP53 野生型和敲除 RPE1 细胞中诱导 WGD;h,YAP1SSA 过表达在 FAT1 野生型和敲除 PC9 细胞中诱导 WGD。
#06 FAT1 缺失加速肿瘤进化和奥希替尼耐药
在奥希替尼耐药的 PC9 细胞模型中,FAT1 敲除细胞表现出更高的克隆存活率、更多的耐药克隆衍生物,且耐药克隆的倍性显著增高。这提示 FAT1 缺失所驱动的 CIN 和 WGD 可能为肿瘤提供进化优势,促进靶向治疗耐药的产生。

图 8 FAT1 缺失导致核形变并加剧靶向治疗耐药。说明:a,PC9 细胞中 FAT1 敲降导致核形态异常(核形态测量);b,活细胞成像显示 FAT1 敲降增加染色质桥形成,且子细胞更易出现核形变;c,初始核形变后,FAT1 敲降细胞后续有丝分裂更易出现异常;d,FAT1 敲降加速有丝分裂进入时间;e,PC9 细胞奥希替尼 IC90 测定;f,耐药克隆生成流程图;g,h,FAT1 敲除增加克隆存活和长期耐药克隆衍生;i,耐药克隆中 FAT1 敲除细胞显示显著变异的 DNA 倍性。
五、结论与研究意义
本研究通过系统的基因组筛选和深入的机制研究,首次揭示了 FAT1 在 NSCLC 中作为一个关键的“双通路”调控因子:一方面,FAT1 缺失通过 YAP1–TEAD 信号轴导致胞质分裂失败和 WGD;另一方面,FAT1 缺失以 YAP1 非依赖的方式损害同源重组修复,导致复制压力增加和结构性 CIN。这两种机制共同放大了肿瘤内异质性,并可能加速肿瘤进化与治疗耐药。该研究不仅为肺鳞癌的早期发生提供了新的分子解释,也提示 Hippo 通路中的 YAP1/TEAD 可能成为克服 FAT1 突变相关耐药的治疗靶点。此外,研究中鉴定的其他五个 DDR/CIN 相关基因(BAP1、CREBBP、NCOA6、RAD21、UBR5)也为未来基于 CIN 和 HRD 的生物标志物开发奠定了基础。
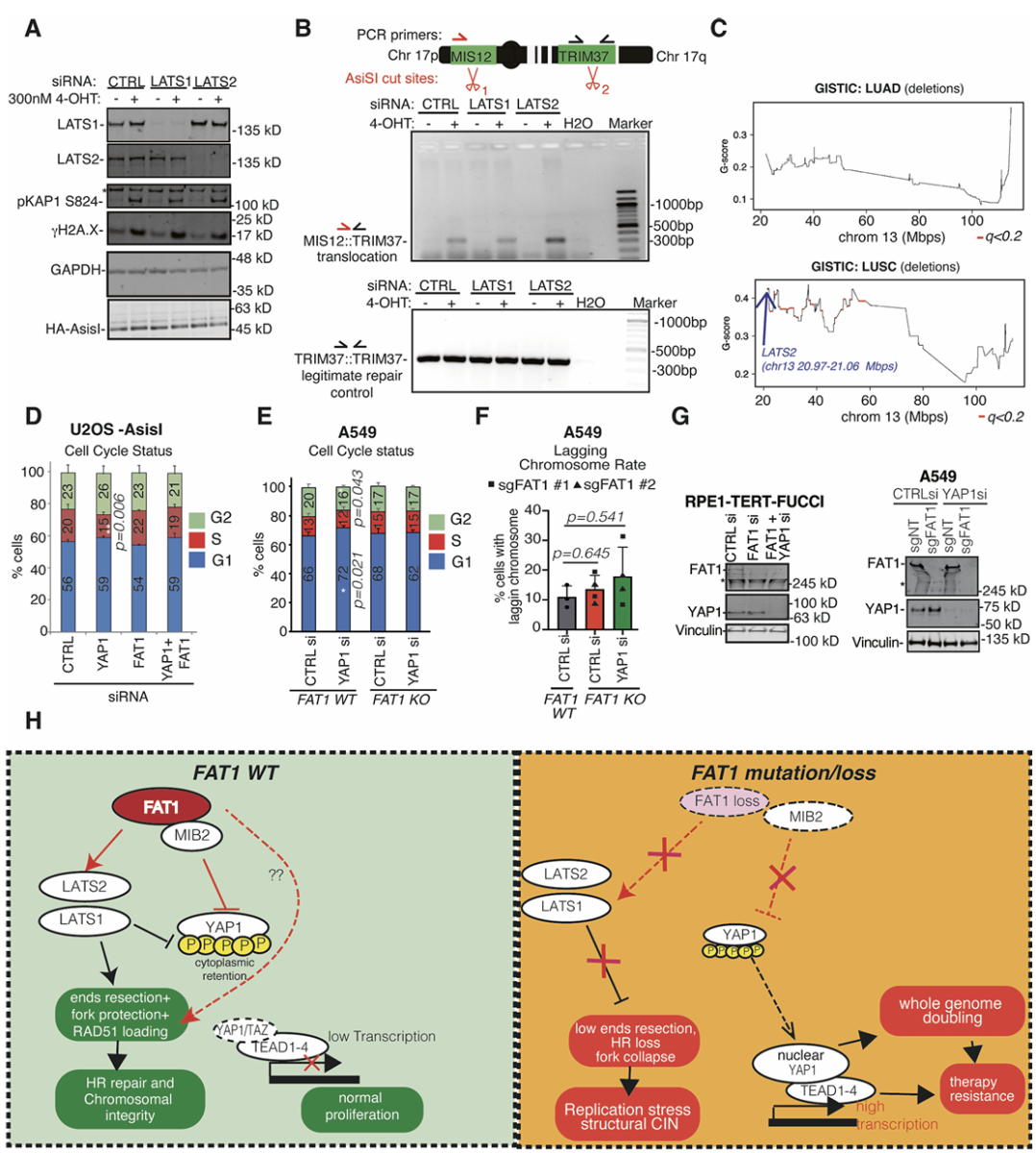
图 9 LATS2 缺失在 TRACERx LUSC 中富集及 FAT1-Hippo 通路模型。说明:a,LATS1/2 敲降后 DNA 损伤标志物正常激活;b,LATS2 敲降诱导不合法修复产物;c,LATS2 基因组位点缺失在 LUSC 中被正选择;d,e,细胞周期分析显示 FAT1/YAP1 共敲除逆转 YAP1 单敲除引起的周期阻滞;f,YAP1 敲降不改变 FAT1 敲除细胞中滞后染色体比例;g,Western blot 验证敲降效率;h,提出的模型:FAT1 通过 YAP1 依赖性通路导致胞质分裂失败和 WGD,通过 YAP1 非依赖性通路导致 HR 缺陷和结构性 CIN。
了解更多 Confocal.nl 产品详情、应用案例或预约 demo,欢迎扫描下方二维码填写信息。

咨询热线:400 857 8882

如果您想要了解更多产品信息,请填写以下信息下载产品手册, 我们收到您的信息后将第一时间回复您。